protein language models
To run the analyses in this chapter, you will need three things.
- Please ensure that your computer can run the following R script. It may prompt you to install additional R packages.
source("https://thebustalab.github.io/phylochemistry/modules/language_model_analysis.R")
## Loading language model module...
## Done with language model loading!- Please create an account at and obtain an API key from https://biolm.ai/ (Login > Account > API Tokens)
- Please create an account (you may also need to create an NVIDIA cloud account if prompted) at and obtain an API key from https://build.nvidia.com/. (To get API key, go to: https://build.nvidia.com/meta/esm2-650m, switch “input” to python and click “Get API Key” > Generate Key)
Keep your API keys (long sequences of numbers and letters, like a password) handy for use in these analyses.
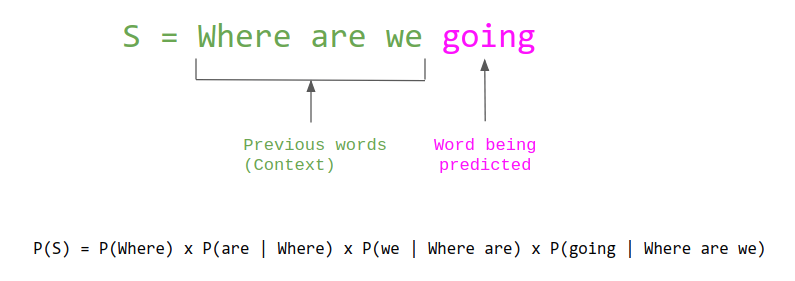
In the last chapter, we saw how language models can convert text into numerical vectors, allowing us to apply statistical and machine learning methods to language data. The same fundamental idea applies to biological sequences. Protein language models are trained on vast databases of protein sequences and learn to convert amino acid sequences into numerical embeddings that capture biologically meaningful patterns — including evolutionary relationships, structural features, and functional properties.
pre-reading
Please read over the following:
- ESM3: Simulating 500 million years of evolution with a language model. The 2024 blog article “ESM3: Simulating 500 million years of evolution with a language model” by EvolutionaryScale introduces ESM3, a revolutionary language model trained on billions of protein sequences. This article explores how ESM3 marks a major advancement in computational biology by enabling researchers to reason over protein sequences, structures, and functions. With massive datasets and powerful computational resources, ESM3 can generate entirely new proteins, including esmGFP, a green fluorescent protein that differs significantly from known natural natural variants. The article highlights the model’s potential to transform fields like medicine, synthetic biology, and environmental sustainability by making protein design programmable. Please note the “Open Model” section of the blog, which highlights applications of ESM models in the natural sciences.
protein embeddings
Autoencoders can be trained to accept various types of inputs, such as text (as shown above), images, audio, videos, sensor data, and sequence-based information like peptides and DNA. Protein language models convert protein sequences into numerical representations that can be used for a variety of downstream tasks, such as structure prediction or function annotation. Protein language models, like their text counterparts, are trained on large datasets of protein sequences to learn meaningful patterns and relationships within the sequence data.
Protein language models offer several advantages over traditional approaches, such as multiple sequence alignments (MSAs). One major disadvantage of MSAs is that they are computationally expensive and become increasingly slow as the number of sequences grows. While language models are also computationally demanding, they are primarily resource-intensive during the training phase, whereas applying a trained language model is much faster. Additionally, protein language models can capture both local and global sequence features, allowing them to identify complex relationships that span across different parts of a sequence. Furthermore, unlike MSAs, which rely on evolutionary information, protein language models can be applied to proteins without homologous sequences, making them suitable for analyzing sequences where little evolutionary data is available. This flexibility broadens the scope of proteins that can be effectively studied using these models.
Beyond the benefits described above, protein language models have an additional, highly important capability: the ability to capture information about connections between elements in their input, even if those elements are very distant from each other in the sequence. This capability is achieved through the use of a model architecture called a transformer, which is a more sophisticated version of an autoencoder. For example, amino acids that are far apart in the primary sequence may be very close in the 3D, folded protein structure. Proximate amino acids in 3D space can play crucial roles in protein stability, enzyme catalysis, or binding interactions, depending on their spatial arrangement and interactions with other residues. Embedding models with transformer architecture can effectively capture these functionally important relationships.
By adding a mechanism called an “attention mechanism” to an autoencoder, we can create a simple form of a transformer. The attention mechanism works within the encoder and decoder, allowing each element of the input (e.g., an amino acid) to compare itself to every other element, generating attention scores that weigh how much attention one amino acid should give to another. This mechanism helps capture both local and long-range dependencies in protein sequences, enabling the model to focus on important areas regardless of their position in the sequence. Attention is beneficial because it captures interactions between distant amino acids, weighs relationships to account for protein folding and interactions, adjusts focus across sequences of varying lengths, captures different types of relationships like hydrophobic interactions or secondary structures, and provides contextualized embeddings that reflect the broader sequence environment rather than just local motifs. For more on attention mechanisms, check out the further reading section of this chapter.
In this section, we will explore how to generate embeddings for protein sequences using a pre-trained protein language model and demonstrate how these embeddings can be used to analyze and visualize protein data effectively. First, we need some data. You can use the OSC_sequences object provided by the source() code, though you can also use the searchNCBI() function to retrieve your own sequences. For example:
g1 <- searchNCBI(search_term = "diterpene synthase", retmax = 10)
g1@ranges@NAMES <- paste0(gsub(" .*", "", g1@ranges@NAMES), "__diterpene_synthase")
g2 <- searchNCBI(search_term = "monoterpene synthase", retmax = 10)
g2@ranges@NAMES <- paste0(gsub(" .*", "", g2@ranges@NAMES), "__monoterpene_synthase")
all_sequences <- c(g1, g2)
all_sequences <- all_sequences[all_sequences@ranges@width < 1024]Once you have some sequences, we can embed them with the function embedAminoAcids(). An example is below. Note that we need to provide either a biolm API key or an NVIDIA api key, and specify which platform we wish to use. We also need to provide the amino acid sequences as an AAStringSet object. If you use the NVIDIA platform, the model esm2-650m will be used (note: esm2 truncates sequences longer than 1022 AA in length). If you use bioLM, you can pick between a number of models.
all_sequences_embedded <- embedAminoAcids(
amino_acid_stringset = all_sequences,
biolm_api_key = readLines("/Users/bust0037/Documents/Websites/biolm_api_key.txt"),
nvidia_api_key = readLines("/Users/bust0037/Documents/Websites/nvidia_api_key.txt"),
platform = "nvidia"
)
all_sequences_embedded$product <- tolower(gsub(".*_", "", all_sequences_embedded$name))
all_sequences_embedded <- select(all_sequences_embedded, name, product, everything())
saveRDS(all_sequences_embedded, "../data/protein_embeddings_example.rds")
all_sequences_embedded[1:5,1:6]Nice! Once we’ve got the embeddings, we can run a PCA analysis to visualize them in 2D space:
all_sequences_embedded_pca <- runMatrixAnalysis(
data = all_sequences_embedded,
analysis = "pca",
columns_w_values_for_single_analyte = colnames(all_sequences_embedded)[3:dim(all_sequences_embedded)[2]],
columns_w_sample_ID_info = c("name", "product")
)
all_sequences_embedded_pca$name <- gsub(".*__", "", all_sequences_embedded_pca$name)
ggplot(all_sequences_embedded_pca) +
geom_jitter(
aes(x = Dim.1, y = Dim.2, fill = name),
shape = 21, size = 5, height = 2, width = 2, alpha = 0.6
) +
scale_fill_manual(values = c("maroon", "gold")) +
theme_minimal()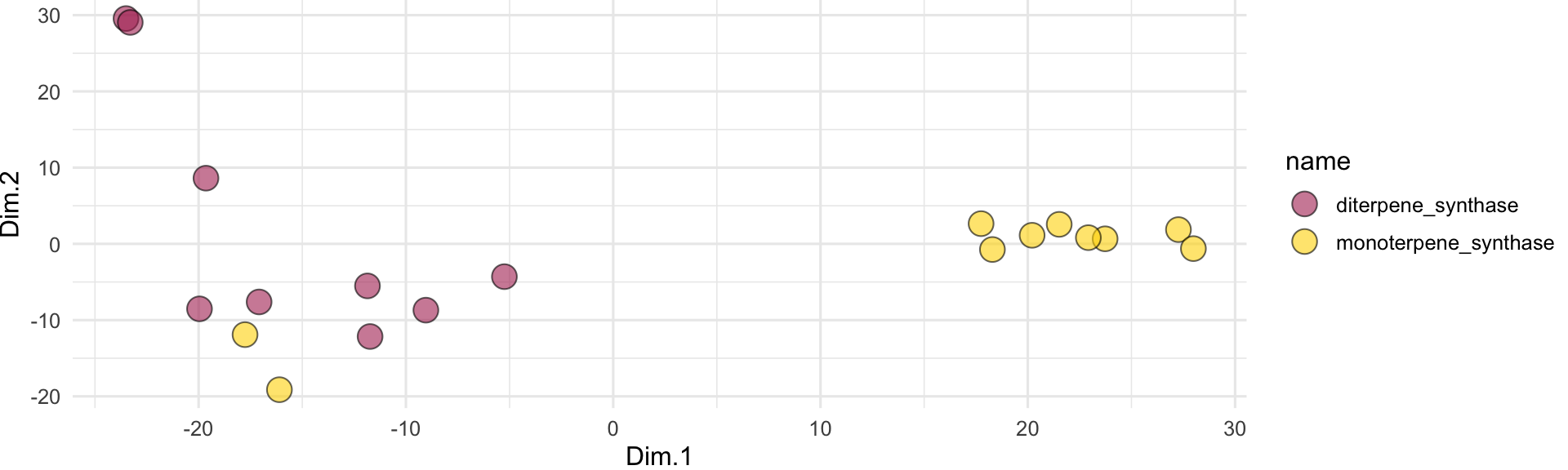
further reading
using protein embeddings in biochemical research. This study presents a machine learning pipeline that successfully identifies and characterizes terpene synthases (TPSs), a challenging task due to the limited availability of labeled protein sequences. By combining a curated TPS dataset, advanced structural domain segmentation, and language model techniques, the authors discovered novel TPSs, including the first active enzymes in Archaea, significantly improving the accuracy of substrate prediction across TPS classes.
attention mechanisms and transformers explained. This Financial Times article explains the development and workings of large language models (LLMs), emphasizing their foundation on the transformer model created by Google researchers in 2017. These models use self-attention mechanisms to understand context, allowing them to respond to subtle relationships between elements in their input, even if those elements are far from one another in the linear input sequence.
other types of protein language models. 3D Protein Structure Prediction deepmind / alphafold2-multimer: Predicts the 3D structure of protein complexes from amino acid sequences. deepmind / alphafold2: Predicts the 3D structure of single proteins from amino acid sequences. meta / esmfold: Predicts the 3D structure of proteins based on amino acid sequences. Protein Embedding Generation meta / esm2-650m: Generates protein embeddings from amino acid sequences. Protein Sequence Design ipd / proteinmpnn: Predicts amino acid sequences for given protein backbone structures. Generative Protein Design ipd / rfdiffusion: A generative model for designing protein backbones, particularly for protein binder design. Molecule-Protein Interaction Prediction mit / diffdock: Predicts the 3D interactions between molecules and proteins (docking simulations).