hierarchical clustering

“Which of my samples are most closely related?”
clustering
So far we have been looking at how to plot raw data, summarize data, and reduce a data set’s dimensionality. It’s time to look at how to identify relationships between the samples in our data sets. For example: in the Alaska lakes dataset, which lake is most similar, chemically speaking, to Lake Narvakrak? Answering this requires calculating numeric distances between samples based on their chemical properties. For this, the first thing we need is a distance matrix:
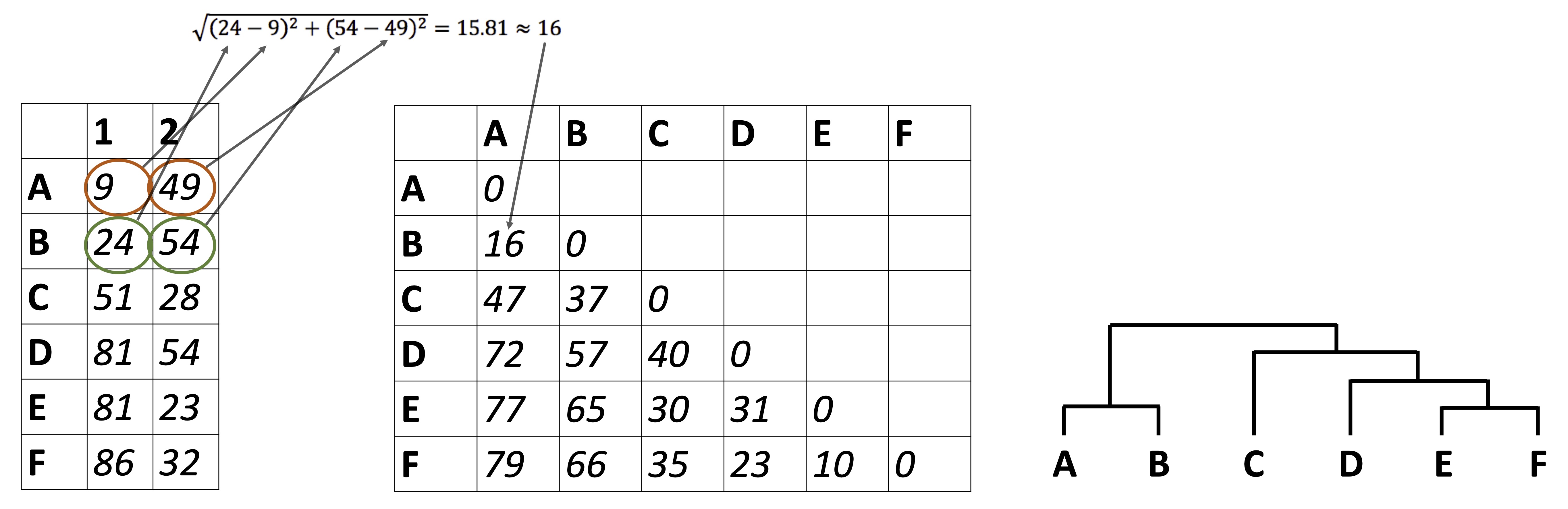
Please note that we can get distance matrices directly from runMatrixAnalyses by specifying analysis = "dist":
alaska_lake_data %>%
select(-element_type) %>%
pivot_wider(names_from = "element", values_from = "mg_per_L") -> alaska_lake_data_wide
dist <- runMatrixAnalyses(
data = alaska_lake_data_wide,
analysis = c("dist"),
columns_w_values_for_single_analyte = colnames(alaska_lake_data_wide)[3:15],
columns_w_sample_ID_info = c("lake", "park")
)
## Replacing NAs in your data with mean
as.matrix(dist)[1:3,1:3]
## sample_unique_ID_sample_1 sample_unique_ID_sample_2
## [1,] "Devil_Mountain_Lake_BELA" "Imuruk_Lake_BELA"
## [2,] "Devil_Mountain_Lake_BELA" "Kuzitrin_Lake_BELA"
## [3,] "Devil_Mountain_Lake_BELA" "Lava_Lake_BELA"
## distance
## [1,] " 3.672034"
## [2,] " 1.663147"
## [3,] " 3.401828"There is more that we can do with distance matrices though, lots more. Let’s start by looking at an example of hierarchical clustering. For this, we just need to tell runMatrixAnalyses() to use analysis = "hclust":
AK_lakes_clustered <- runMatrixAnalyses(
data = alaska_lake_data_wide,
analysis = c("hclust"),
columns_w_values_for_single_analyte = colnames(alaska_lake_data_wide)[3:15],
columns_w_sample_ID_info = c("lake", "park")
)
AK_lakes_clustered
## # A tibble: 38 × 26
## sample_unique_ID lake park parent node branch.length
## <chr> <chr> <chr> <int> <int> <dbl>
## 1 Devil_Mountain_La… Devi… BELA 23 1 7.81
## 2 Imuruk_Lake_BELA Imur… BELA 25 2 6.01
## 3 Kuzitrin_Lake_BELA Kuzi… BELA 24 3 3.27
## 4 Lava_Lake_BELA Lava… BELA 32 4 3.14
## 5 North_Killeak_Lak… Nort… BELA 38 5 256.
## 6 White_Fish_Lake_B… Whit… BELA 38 6 0.828
## 7 Iniakuk_Lake_GAAR Inia… GAAR 36 7 2.59
## 8 Kurupa_Lake_GAAR Kuru… GAAR 30 8 6.00
## 9 Lake_Matcharak_GA… Lake… GAAR 35 9 2.09
## 10 Lake_Selby_GAAR Lake… GAAR 29 10 3.49
## # ℹ 28 more rows
## # ℹ 20 more variables: label <chr>, isTip <lgl>, x <dbl>,
## # y <dbl>, branch <dbl>, angle <dbl>, bootstrap <lgl>,
## # water_temp <dbl>, pH <dbl>, C <dbl>, N <dbl>, P <dbl>,
## # Cl <dbl>, S <dbl>, F <dbl>, Br <dbl>, Na <dbl>,
## # K <dbl>, Ca <dbl>, Mg <dbl>It works! Now we can plot our cluster diagram with a ggplot add-on called ggtree. We’ve seen that ggplot takes a “data” argument (i.e. ggplot(data = <some_data>) + geom_*() etc.). In contrast, ggtree takes an argument called tr, though if you’re using the runMatrixAnalysis() function, you can treat these two (data and tr) the same, so, use: ggtree(tr = <output_from_runMatrixAnalyses>) + geom_*() etc.
Note that ggtree also comes with several great new geoms: geom_tiplab() and geom_tippoint(). Let’s try those out:
library(ggtree)
AK_lakes_clustered %>%
ggtree() +
geom_tiplab() +
geom_tippoint() +
theme_classic() +
scale_x_continuous(limits = c(0,700))
Cool! Though that plot could use some tweaking… let’s try:
AK_lakes_clustered %>%
ggtree() +
geom_tiplab(aes(label = lake), offset = 10, align = TRUE) +
geom_tippoint(shape = 21, aes(fill = park), size = 4) +
scale_x_continuous(limits = c(0,600)) +
scale_fill_brewer(palette = "Set1") +
# theme_classic() +
theme(
legend.position = c(0.4,0.2)
)
Very nice! Since North Killeak and White Fish are so different from the others, we could re-analyze the data with those two removed:
alaska_lake_data_wide %>%
filter(!lake %in% c("North_Killeak_Lake","White_Fish_Lake")) -> alaska_lake_data_wide_filtered
runMatrixAnalyses(
data = alaska_lake_data_wide_filtered,
analysis = c("hclust"),
columns_w_values_for_single_analyte = colnames(alaska_lake_data_wide_filtered)[3:15],
columns_w_sample_ID_info = c("lake", "park")
) %>%
ggtree() +
geom_tiplab(aes(label = lake), offset = 10, align = TRUE) +
geom_tippoint(shape = 21, aes(fill = park), size = 4) +
scale_x_continuous(limits = c(0,100)) +
scale_fill_brewer(palette = "Set1") +
# theme_classic() +
theme(
legend.position = c(0.05,0.9)
)
## Replacing NAs in your data with mean
Annotating trees
Overlaying sample traits on a ggtree-based plot is straightforward when we combine ggtree with ggplot2. We begin by running the hierarchical clustering analysis and keeping its output for later plotting.
hclust_out <- runMatrixAnalyses(
data = chemical_blooms,
analysis = c("hclust"),
columns_w_values_for_single_analyte = colnames(chemical_blooms)[2:10],
columns_w_sample_ID_info = "label"
)
## ! The tree contained negative edge lengths. If you want to
## ignore the edges, you can set
## `options(ignore.negative.edge=TRUE)`, then re-run ggtree.The object returned by runMatrixAnalyses() already contains the branch coordinates, so we can pass it directly to ggtree() and add tip labels while keeping the tree readable. Note that we should deliberately control the y-axis here, that will be key for aligning the tree with other plots later.
tree_plot <- ggtree(hclust_out) +
geom_tiplab(size = 2, align = TRUE) +
scale_x_continuous(limits = c(0, 300)) +
scale_y_continuous(limits = c(0, 80)) +
theme_classic()
## Scale for y is already present.
## Adding another scale for y, which will replace the existing
## scale.
tree_plot
Next, reshape the tip-level measurements to long form so each chemical becomes its own column of tiles. Because we reuse the y coordinate supplied by ggtree, the tiles inherit the same vertical order as the tips in the tree. Note that we remove the other columns in the hclust output for simplicity - they are only needed if we want to draw the full tree. Note that we also control the y-axis here to make sure it has the same bounds (limits) as the tree we made previously.
heat_plot <- hclust_out %>%
filter(isTip) %>%
select(-parent, -node, -branch.length, -label, -isTip, -x, -branch, -angle, -bootstrap) %>%
pivot_longer(cols = 3:11, names_to = "chemical", values_to = "abundance") %>%
ggplot(aes(x = chemical, y = y, fill = abundance)) +
geom_tile() +
scale_y_continuous(limits = c(0, 80)) +
theme(axis.text.x = element_text(angle = 90, hjust = 1))
heat_plot
With matching y scales, plot_grid() can align the tree and the heat map so the tiles line up with the corresponding samples. Using align = "h" snaps them together horizontally, and axis = "tb" keeps the panel heights consistent.
plot_grid(tree_plot, heat_plot, axis = "tb", align = "h")
Note: if we were to instead build the heat map directly from the raw chemical_blooms table, the rows fall back to their alphabetical order and the heat map no longer matches the dendrogram ordering:
chemical_blooms %>%
pivot_longer(cols = 2:10, names_to = "chemical", values_to = "abundance") %>%
ggplot(aes(x = chemical, y = label, fill = abundance)) +
geom_tile()
further reading
annotating phylogenetic trees with ggtree. This free, book-length reference from the ggtree authors walks through annotating dendrograms and phylogenetic trees in R, explaining how to layer metadata, add tip labels, and customize themes—perfect for polishing the cluster plots we generated in this chapter.
hierarchical clustering in R. UC Business Analytics’ tutorial provides a gentle, R-focused walkthrough of agglomerative clustering, covering distance matrices, linkage choices, dendrogram interpretation, and cluster cutting with reproducible code examples. Note that this text uses base R instead of our in-class functions.