data visualization I
Visualization is one of the most fun parts of working with data. In this section, we will jump into visualization as quickly as possible - after just a few prerequisites. Please note that data visualization is a whole field in and of itself (just google “data visualization” and see what happens). Data visualization is also rife with “trendy” visuals, misleading visuals, and visuals that look cool but don’t actually communicate much information. We will touch on these topics briefly, but will spend most of our time practicing how to represent our data in intuitive and interpretable ways. Let’s get started!
objects
In R, data is stored in objects. You can think of objects as if they were “files” inside an R session. phylochemistry provides a variety of objects for us to work with. Let’s look at how to create an object. For this, we can use an arrow: <- . The arrow will take something and store it inside an object. For example:
new_object <- 1Now we’ve got a new object called new_object, and inside of it is the number 1. To look at what’s inside an object, we can simply type the name of the object into the console:
new_object
## [1] 1Easy! Let’s look at one of the objects that comes with our class code base. What are the dimensions of the “algae_data” data set?
algae_data
## # A tibble: 180 × 5
## replicate algae_strain harvesting_regime chemical_species
## <dbl> <chr> <chr> <chr>
## 1 1 Tsv1 Heavy FAs
## 2 1 Tsv1 Heavy saturated_Fas
## 3 1 Tsv1 Heavy omega_3_polyuns…
## 4 1 Tsv1 Heavy monounsaturated…
## 5 1 Tsv1 Heavy polyunsaturated…
## 6 1 Tsv1 Heavy omega_6_polyuns…
## 7 1 Tsv1 Heavy lysine
## 8 1 Tsv1 Heavy methionine
## 9 1 Tsv1 Heavy essential_Aas
## 10 1 Tsv1 Heavy non_essential_A…
## # ℹ 170 more rows
## # ℹ 1 more variable: abundance <dbl>functions
Excellent - we’ve got data. Now we need to manipulate it. For this we need functions:
- A function is a command that tells R to perform an action!
- A function begins and ends with parentheses:
this_is_a_function() - The stuff inside the parentheses are the details of how you want the function to perform its action:
run_this_analysis(on_this_data)
Let’s illustrate this with an example. algae_data is a pretty big object. For our next chapter on visualization, it would be nice to have a smaller dataset object to work with. Let’s use another tidyverse command called filter to filter the algae_data object. We will need to tell the filter command what to filter out using “logical predicates” (things like equal to: ==, less than: <, greater than: >, greater-than-or-equal-to: <=, etc.). Let’s filter algae_data so that only rows where the chemical_species is equal to FAs (fatty acids) is preserved. This will look like chemical_species == "FAs". Here we go:
filter(algae_data, chemical_species == "FAs")
## # A tibble: 18 × 5
## replicate algae_strain harvesting_regime chemical_species
## <dbl> <chr> <chr> <chr>
## 1 1 Tsv1 Heavy FAs
## 2 2 Tsv1 Heavy FAs
## 3 3 Tsv1 Heavy FAs
## 4 1 Tsv1 Light FAs
## 5 2 Tsv1 Light FAs
## 6 3 Tsv1 Light FAs
## 7 1 Tsv2 Heavy FAs
## 8 2 Tsv2 Heavy FAs
## 9 3 Tsv2 Heavy FAs
## 10 1 Tsv2 Light FAs
## 11 2 Tsv2 Light FAs
## 12 3 Tsv2 Light FAs
## 13 1 Tsv11 Heavy FAs
## 14 2 Tsv11 Heavy FAs
## 15 3 Tsv11 Heavy FAs
## 16 1 Tsv11 Light FAs
## 17 2 Tsv11 Light FAs
## 18 3 Tsv11 Light FAs
## # ℹ 1 more variable: abundance <dbl>Cool! Now it’s just showing us the 18 rows where the chemical_species is fatty acids (FAs). Let’s write this new, smaller dataset into a new object. For that we use <-, remember?
algae_data_small <- filter(algae_data, chemical_species == "FAs")
algae_data_small
## # A tibble: 18 × 5
## replicate algae_strain harvesting_regime chemical_species
## <dbl> <chr> <chr> <chr>
## 1 1 Tsv1 Heavy FAs
## 2 2 Tsv1 Heavy FAs
## 3 3 Tsv1 Heavy FAs
## 4 1 Tsv1 Light FAs
## 5 2 Tsv1 Light FAs
## 6 3 Tsv1 Light FAs
## 7 1 Tsv2 Heavy FAs
## 8 2 Tsv2 Heavy FAs
## 9 3 Tsv2 Heavy FAs
## 10 1 Tsv2 Light FAs
## 11 2 Tsv2 Light FAs
## 12 3 Tsv2 Light FAs
## 13 1 Tsv11 Heavy FAs
## 14 2 Tsv11 Heavy FAs
## 15 3 Tsv11 Heavy FAs
## 16 1 Tsv11 Light FAs
## 17 2 Tsv11 Light FAs
## 18 3 Tsv11 Light FAs
## # ℹ 1 more variable: abundance <dbl>Here are a variety of ways to filter:
filter(<data>, <variable> < 18) ## less than 18
filter(<data>, <variable> <= 18) ## less than or equal to 18
filter(<data>, <variable> > 18) ## greater than 18
filter(<data>, <variable> >= 18) ## greater than or equal to 18
filter(<data>, <variable> == 18) ## equal to 18
filter(<data>, <variable> != 18) ## not equal to 18
filter(<data>, <variable> == 18 | <variable> == 19) ## equal to 18 or 19
filter(<data>, <variable> %in% c(18, 19, 20)) ## equal to 18 or 19 or 20
concept check
Fill in the the blank below so that only rows where chemical_species equals "FAs" are kept, then press Run to see the output of the filter.
Self-check: inside filter(), which operator tests whether two values are equal?
ggplot & geoms
Now we have a nice, small table that we can use to practice data visualization. For visualization, we’re going to use ggplot2 - a powerful set of commands for plot generation.
There are three steps to setting up a ggplot:
- Define the data you want to use.
We do this using the ggplot function’s data argument. When we run that line, it just shows a grey plot space. Why is this? It’s because all we’ve done is told ggplot that (i) we want to make a plot and (ii) what data should be used. We haven’t explained how to represent features of the data using ink.
ggplot(data = algae_data_small)
- Define how your variables map onto the axes.
This is called aesthetic mapping and is done with the aes() function. aes() should be placed inside the ggplot command. Now when we run it, we get our axes!
ggplot(data = algae_data_small, aes(x = algae_strain, y = abundance))
- Use geometric shapes to represent other variables in your data.
Map your variables onto the geometric features of the shapes. To define which shape should be used, use a geom_* command. Some options are, for example, geom_point(), geom_boxplot(), and geom_violin(). These functions should be added to your plot using the + sign. We can use a new line to keep the code from getting too wide, just make sure the + sign is at the end of the top line. Let’s try it:
ggplot(data = algae_data_small, aes(x = algae_strain, y = abundance)) +
geom_point()
In the same way that we mapped variables in our dataset to the plot axes, we can map variables in the dataset to the geometric features of the shapes we are using to represent our data. For this, again, use aes() to map your variables onto the geometric features of the shapes:
ggplot(data = algae_data_small, aes(x = algae_strain, y = abundance)) +
geom_point(aes(color = harvesting_regime))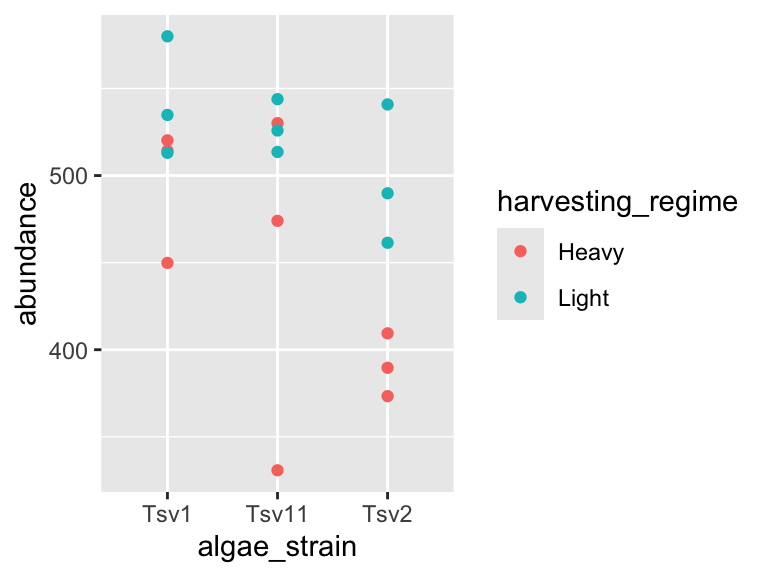
Concept check
First make the small algae table by filling in the blank (first window below), then plot it (second window below). Edit the code and press Run.
Self-check: in the plot above, what does the point color represent?
In the plot above, the points are a bit small, how could we fix that? We can modify the features of the shapes by adding additional arguments to the geom_*() functions. To change the size of the points created by the geom_point() function, this means that we need to add the size = argument. IMPORTANT! Please note that when we map a feature of a shape to a variable in our data (as we did with color/harvesting regime, above) then it goes inside aes(). In contrast, when we map a feature of a shape to a constant, it goes outside aes(). Here’s an example:
ggplot(data = algae_data_small, aes(x = algae_strain, y = abundance)) +
geom_point(aes(color = harvesting_regime), size = 5)
One powerful aspect of ggplot is the ability to quickly change mappings to see if alternative plots are more effective at bringing out the trends in the data. For example, we could modify the plot above by switching how harvesting_regime is mapped:
ggplot(data = algae_data_small, aes(x = algae_strain, y = abundance)) +
geom_point(aes(size = harvesting_regime), color = "black")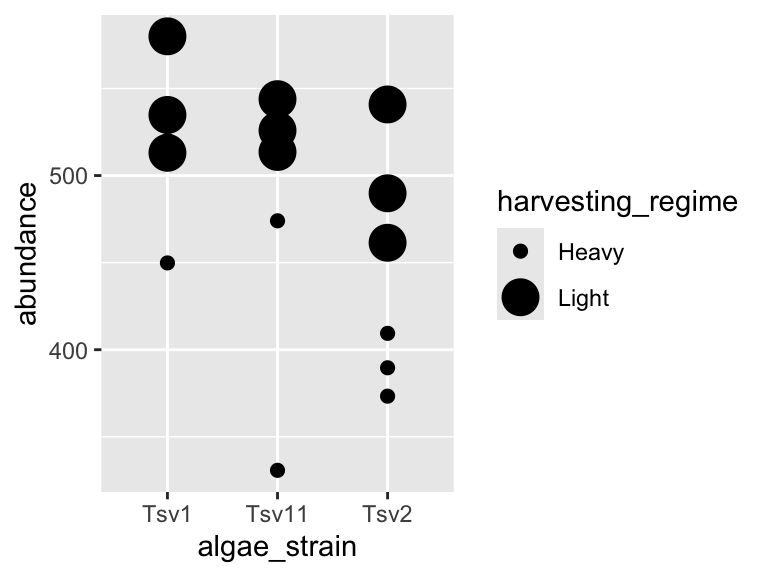
Important note: Inside the aes() function, map aesthetics (the features of the geom’s shape) to a variable. Outside the aes() function, map aesthetics to constants. You can see this in the above two plots - in the first one, color is inside aes() and mapped to the variable called harvesting_regime, while size is outside the aes() call and is set to the constant 5. In the second plot, the situation is reversed, with size being inside the aes() function and mapped to the variable harvesting_regime, while color is outside the aes() call and is mapped to the constant “black”.
We can also stack geoms on top of one another by using multiple + signs. We also don’t have to assign the same mappings to each geom.
ggplot(data = algae_data_small, aes(x = algae_strain, y = abundance)) +
geom_violin() +
geom_point(aes(color = harvesting_regime), size = 5)
As you can probably guess right now, there are lots of mappings that can be done, and lots of different ways to look at the same data!
ggplot(data = algae_data_small, aes(x = algae_strain, y = abundance)) +
geom_violin(aes(fill = algae_strain)) +
geom_point(aes(color = harvesting_regime, size = replicate))
ggplot(data = algae_data_small, aes(x = algae_strain, y = abundance)) +
geom_boxplot()
exercises


further reading
R for Data Science: Data transformation. The dplyr chapter of Wickham and Grolemund’s book, covering
filter,select, and the other core verbs for reshaping data before plotting.dplyr cheat sheet. A one-page visual reference for the dplyr data-manipulation verbs, handy to keep beside you while filtering.
ggplot2 cheat sheet. A compact overview of the ggplot2 grammar (geoms, aesthetics, facets), useful for recalling syntax at a glance.
R Markdown: The Definitive Guide. Xie, Allaire, and Grolemund’s reference for authoring reproducible reports, covering chunk options, output formats, and document structure.
R Markdown cheat sheet. A two-page summary of R Markdown syntax and knitr chunk options for quickly formatting reports.